„Szilárdtestfelületek analízise Auger elektron spektroszkópiával” változatai közötti eltérés
Lenk (vitalap | szerkesztései) |
Lenk (vitalap | szerkesztései) |
||
| 95. sor: | 95. sor: | ||
{{eq|I_i(WXY) {{=}} I_p \cdot s_i \cdot N \cdot X_i|eq:1|(1)}} | {{eq|I_i(WXY) {{=}} I_p \cdot s_i \cdot N \cdot X_i|eq:1|(1)}} | ||
Mint láttuk s<sub>i</sub> nagyon sok, néha csak nagy bizonytalansággal számolható, mennyiségtől függ. Azonban néhány megszorító feltételezés bevezetésével bizonyos mintatípusoknál a mennyiségi meghatározás módja leegyszerűsíthető. Így eltekintünk a felületi érdrsség befolyásoló szerepétől. (Az ebből származó hibát polírozott, vagy sík szemcsehatár felületek előállításával, illetőleg forgatható mintatartó alkalmazásával lehet csökkenteni.) Továbbá figyelembe vesszük, hogy a nem kifejezetten kémiai vegyületeknél a mátrix környezet elsősorban az elemek legkülső elektronhéjainak energiaviszonyait befolyásolja, és a mélynívókra gyakorolt hatása sokkal kisebb jelentőségű. (Így a kiértékelésnél csak azokat az energia csúcsokat használjuk fel, amelyeknek kialakításában a mélynívók vesznek részt.). Az s<sub>i</sub> mátrix függése még tovább csökkenthető, ha relatív érzékenységi tényezőkkel (RSF) dolgozunk, azaz egy önkényesen megválasztott anyag (általában ezüst) érzékenységi faktorához viszonyítjuk (S<sub>i</sub>=s<sub>i</sub>/s<sub>Ag</sub>). Egy adott berendezés típusra valamennyi tiszta anyag relatív érzékenységi tényezője kimérhető és a fent ismertetett feltételek teljesülése esetén jó közelítéssel használható ismeretlen összetételű anyagokra is. [[#eq:1|(1)]] összefüggésből az adott elem koncentrációja (atomtörtben) kifejezve: | Mint láttuk s<sub>i</sub> nagyon sok, néha csak nagy bizonytalansággal számolható, mennyiségtől függ. Azonban néhány megszorító feltételezés bevezetésével bizonyos mintatípusoknál a mennyiségi meghatározás módja leegyszerűsíthető. Így eltekintünk a felületi érdrsség befolyásoló szerepétől. (Az ebből származó hibát polírozott, vagy sík szemcsehatár felületek előállításával, illetőleg forgatható mintatartó alkalmazásával lehet csökkenteni.) Továbbá figyelembe vesszük, hogy a nem kifejezetten kémiai vegyületeknél a mátrix környezet elsősorban az elemek legkülső elektronhéjainak energiaviszonyait befolyásolja, és a mélynívókra gyakorolt hatása sokkal kisebb jelentőségű. (Így a kiértékelésnél csak azokat az energia csúcsokat használjuk fel, amelyeknek kialakításában a mélynívók vesznek részt.). Az s<sub>i</sub> mátrix függése még tovább csökkenthető, ha relatív érzékenységi tényezőkkel (RSF) dolgozunk, azaz egy önkényesen megválasztott anyag (általában ezüst) érzékenységi faktorához viszonyítjuk (S<sub>i</sub>=s<sub>i</sub>/s<sub>Ag</sub>). Egy adott berendezés típusra valamennyi tiszta anyag relatív érzékenységi tényezője kimérhető és a fent ismertetett feltételek teljesülése esetén jó közelítéssel használható ismeretlen összetételű anyagokra is. [[#eq:1|(1)]] összefüggésből az adott elem koncentrációja (atomtörtben) kifejezve: | ||
| − | $$X_i {{=}} \frac{I_i(WXY)/s_i}{\ | + | $$X_i {{=}} \frac{I_i(WXY)/s_i}{\sum_{k} I_k(WXY)/s_k}$$ |
| − | + | ahol a k szerinti összegzés a minta összes detektálható elemére terjed ki. Végigosztva a számlálót is és a nevezőt is s<sub>Ag</sub>-vel: | |
| − | + | $$X_i {{=}} \frac{I_i(WXY)/S_i}{\sum_{k} I_k(WXY)/S_k}$$ | |
| + | Ez az összefüggés 10-15% pontossággal alkalmazható nem kémiai vegyületek összetételének meghatározására. A módszer érzékenysége 1-5 at%, ami egyéb kémiai analízissel összehasonlítva igen szerénynek tűnhet. Azonban vegyük figyelembe, hogy az a térfogat amiből az információ származik néhány tized nm vastag réteg, közepesen jól fókuszált primer elektronnyaláb (átmérő: $\sim 1\mu$ esetében, ~10<sup>-8</sup> cm2 területű tartományából. Az atomi sűrűséget 1023 atom/cm<sup>3</sup>-nek véve 1% érzékenységgel számolva ez néhányszor 10<sup>4</sup> atom kimutatását jelenti. | ||
| + | ===Kötésállapot kimutatása=== | ||
| + | Az Auger mérésekből nyerhető másik fontos információ a kémiai kötésviszonyokra levonható következtetés. Erre az ad lehetőséget, hogy a különböző kémiai környezet | ||
A lap 2012. november 20., 17:34-kori változata
Tartalomjegyzék[elrejtés] |
Szerkesztés alatt!
Elméleti összefoglaló
Az AES módszer elve
Pierre Auger francia fizikus röntgensugárzással gerjesztett argon atomok gerjesztési folyamatainak Wilson-féle ködkamrában történő tanulmányozása folyamán fedezte fel a róla elnevezett effektust 1925-ben. Ezt követően csak kb. 40 év után kezdődött el az AES módszer széleskörű gyakorlati felhasználása. Napjainkban az AES módszert elsősorban szilárdtest felületek vizsgálatára alkalmazzák. Ennek folyamán a vizsgálandó felületet valamilyen primer gerjesztés hatásának tesszük ki, ami elsősorban 1-10 keV-os elektronnyalábbal való bombázást jelent, de elektromágneses sugárzás vagy ionnyaláb is lehet a gerjesztő hatás. Az ekkor keletkező un. Auger elektronok energiáinak mérésével lehet a minta összetételét meghatározni.
A vizsgálandó anyagban a primer gerjesztés hatására végbemenő Auger-folyamatot az 1. ábrán szemléltetjük, és az alábbiak szerint értelmezzük.
 |
Egy belső, például a K-héjon lévő elektront a primer részecske eltávolít az atomi kötelékből. Az így szabaddá váló energianívóra egy magasabb, például az L1 nívóról lép be egy elektron. A felszabaduló energiát például az L2 nívón lévő elektron veszi át, ami egy jól meghatározott, karakterisztikus energiával kilép a felületből. Ezt az elektront nevezzük KL1L2, vagy általánosabban KLL Auger-elektronnak. Itt jegyezzük meg, hogy a gyakorlat számára legfontosabb, nagy elektronhozammal rendelkező Auger-elektronok esetén az Auger-folyamatban résztvevő második és harmadik elektronhéj egybeesik, így az anyagok zöménél a KLL, LMM vagy MNN Auger-elektronok a leggyakoriabbak.
A továbbiakban határozzuk meg, az atomból emittálódó Auger-elektron energiáját nullad rendű közelítésben. Legyenek EK, EL1 és EL2 a megfelelő héjakhoz tartozó ionizációs energiák. Amikor az L1 héjon lévő elektron betölti a K héjon lévő üres helyet 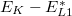 energia szabadul. (A csillag felső index azt jelzi, hogy az adott atomi nivó egy gerjesztett atom energia nívója.) Igy az L2 nivóról távozó Auger elektron mozgási energiája:
energia szabadul. (A csillag felső index azt jelzi, hogy az adott atomi nivó egy gerjesztett atom energia nívója.) Igy az L2 nivóról távozó Auger elektron mozgási energiája:
![\[E_{KLL} {{=}} E_K-E_{L1}^* - E_{L2}^*\]](/images/math/c/a/a/caa7e200dcc4c72449cd59a5d5a4f260.png)
Nullad rendű közelítésben az egyes ionizációs energiákat az adott atomi nívók energiáival azonosíthatjuk. Azonban ez nem veszi figyelembe, hogy az Auger folyamatban egyszeresen és kétszeresen ionizált állapotú atomok szerepelnek, továbbá azt sem hogy az elektronok szilárd testetből származnak. Mindezeket figyelembe vevő számítások bonyolultak, viszont kisérletileg jól kimérhetőek az egyes elemekre jellemző Auger átmenetek energiái. Ezekről ad áttekintést a 2. ábra.
 |
Megjegyezzük még, hogy ha az Auger-átmenetben a valencia-sáv is részt vesz, akkor az Auger elektron energiák, illetve ezek eltolódásainak mérésével következtetéseket lehet levonni a mintát alkotó atomok kötésviszonyairól is.
AES berendezések felépítése
Az AES mérések ultravákuum körülményeket igényelnek. Erről a felületanalitikai mérési módszerek általános bevezetőjében beszeltünk. Az AES berendezések két fő egysége a primer elektron forrás és az energia analizátor a detektorral.
Primer elektron forrás
Auger elektronokat többféle módon is ki lehet váltani, például röntgen sugárzással, elektron vagy ionbombázással. Gyakorlatban azonban az elektronbombázásos gerjesztés terjedt el. Ennek fő előnye, hogy viszonylag könnyű és olcsó létrehozni megfelelő energiájú és intenzitású elektronokat. Az elektronsugár elektrosztatikusan jól fókuszálható és mozgatható. Jól fókuszált elektronnyaláb esetén a minta szekunder elektron képét is elő lehet állítani, ami lehetővé teszi a minta tetszőleges pontjának kémiai analízisét. Hasonló módon így arra is van mód, hogy elemeloszlás térképet is vegyünk fel. Az ilyen berendezések szokásos elnevezése SAM (Scanning Auger Microprobe). Az Auger berendezésekben használt ionágyúk általában 0-10 keV energiájú elektronok előállítására alkalmasak. Általában 3-5 keV energiájú elektronokkal lehet a legnagyobb intenzitású Auger áramot gerjeszteni, de a jobb fókuszálhatóság érdekében sokszor nagyobb energiákat is használnak. A gerjesztő nyaláb intenzitása a megfelelő érzékenység elérése érdekében 10-6-10-9 A kell legyen.
Energia analizátor
Az elektronok energia szerinti analízisét elektrosztatikus eltérítésű analizátorokkal lehet elvégezni. Az analizátorban az elektromos tér az elektronokat mozgási energiájuk függvényében más és más pályára kényszeríti, és ez teszi lehetővé szeparációjukat. Az energiafelbontást (tehát azt, hogy milyen energiakülönbségű elektronokat tudunk egymástól elkülöníteni) az analizátor típusa és műszaki paraméterei (pl. mérete) határozzák meg.
Gyakran használják a CMA (hengeres tükör) analizátorokat, melyekben az elektronok szétválasztása két koaxiális henger közötti logaritmikus elektromos térben zajlik. CMA-val jó intenzitású, de gyengébb felbontású spektrumok vehetők fel. A CMA-nál a minta elhelyezésére szigorú geometriai megszorítások vonatkoznak.
A koncentrikus félgömb (vagy hemiszférikus) analizátorok (CHA) jobb felbontást ad az elérhető intenzitás viszont romlik.
Az energia analizátoron másodpercenként <106 elektron jut át. Ilyen kis áramok (<10-13 A) detektálásához elktronsokszorozókat (manapság ugynevezett chaneneltronokat) alkalmaznak. Ezekkel ~107-szeres erősítést lehet elérni.
Az AES méréseknél használt készülék műszaki adatai
A laborméréseket a BME Atomfizika Tanszékén található berendezésen végezzük. A Budapesti Műszaki Egyetem Atomfizika Tanszékének Felületfizikai Laboratóriumában egy VG Microtech gyártmányú XPS-SAM komplex felületanalitikai nagyműszer működik, melyben XPS és Auger elektron spektroszkópiai vizsgálatok is végezhetőek.
A berendezésben használt elektronágyú VG Microtech gyártmányú, típusa: LEG200. Minimális nyalábátmérő: 200nm, energia tartomány: 0-10keV, maximális áram: 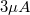 .
.
Az elektron energia analizátor egy CLAM 2 típusú, VG gyártmányú, csonkított hemiszférikus, 180°-os szektoranalizátor. A hozzá kapcsolódó detektor 106-108-szoros erősítésű channeltron.
A minták szekunder elektron képét egy szcintillációs számláló segítségével lehet előállítani. A minta felületéről kilépő elektronok (kinetikus energiájuktól függetlenül) egy a vákuumtérbe merülő olyan bevonattal ellátott felületbe ütköznek, melyben fényfelvillanásokat okoznak. A fényfelvillanásokat fotoelektron-sokszorózóval detektáljuk. Az így keletkező jelet szinkronba hozzuk a primer elektron–forrás pásztázásával. A keletkező kép különböző fényességű területei a minta megfelelő részeiről kilépő elektronok számát jellemzi.
A minták felületének in-situ tisztításához az Atomfizika Tanszéken fejlesztett, differenciálisan szívott, elektronütközéses argon ionágyút használunk. A tipikus porlasztó ionáram 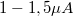 , a porlasztott terület nagysága ionoptika segítségével
, a porlasztott terület nagysága ionoptika segítségével 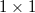 -től
-től 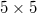 mm-ig változtatható.
mm-ig változtatható.
Az analitikai kamrában a vákuumot egy VEB HD EGZ 250.8 iongetter- és egy Edwards gyártmányú EXT 250 turbomolekuláris szivattyú biztosítja. Az iongetter szivattyú szívássebessége levegőre 560 l/s, argonra 152 l/s. A turbomolekuláris szivattyú szívássebessége nitrogénre 240 l/sec, nemesgázokra 250 l/sec. A mérési pozíció megfelelő megválasztását a mintatartó manipulátor teszi lehetővé, amely a 3 térbeli irányban (x,y és z mentén) elmozdítható, és a tartószerkezet vákuumtérbe benyúló tengelye körül elforgatható. A beállítás az x,y,z tengelyeken 0,01mm-, a forgatásnál 1° pontossággal kivitelezhető. A minták in-situ 1000°C-ig fűthetőek. A mintákat egy EXT 250 típusú turbomolekuláris szivattyúval szívott zsilipkamrán keresztül juttatjuk az analitikai kamrába.
A mérés-adatgyűjtés a VGX900 nevű szoftver segítségével automatizált. A detektorok által észlelt jelet megfelelő erősítés után a számítógép regisztrálja. A számítógép lépteti megadott diszkrét értékekkel a spektrométer energia-analizátorát, és lépésenként tárolja a jelintenzitást. A program rendelkezik adatfeldolgozási funkciókkal is.
AES spektrumok és kiértékelésük
A 3. ábrán szén mintán mért szekunder elektron eloszlás látható az energia függvényében. A mérés során a gerjesztő primer elektronok energiája 1000 eV volt. A spektrumon ennél az energiánál jelentkező nagy csúcs a rugalmasan visszaszórt elektronok következménye. A direkt spektrumon (N(E)) jól látható, hogy az Auger csúcsok egy igen nagy háttéren ülnek. Ez az Auger mérések nagy problémája. Ettől a folytonos lassan változó háttértől deriválással szabadulhatunk meg. A 3. ábrán a dN(E)/dE görbén már jól meghatározható helyen jelentkezik a C (KLL) csúcsa és a jel csúcstól csúcsig mért értéke is könnyen mérhető. A gyakorlatban az Auger csúcs nagyságát ezen csúcstól csúcsig mért távolsággal adjuk meg. Manapság az N(E) direkt spektrumból a dN(E)/dE spektrumot számítógéppel numerikus deriválással határozzák meg.
 |
Kvalitatív analízis
Igen sok esetben csak arra van szükségünk, hogy eldöntsük egy adott felületen bizonyos kezelések, eljárások után van-e valamilyen elem. Ezekben az esetekben csak az a feladat, hogy elegendően széles energia tartományban nagy érzékenységgel vegyünk fel Auger elektron spektrumokat. A mért csúcsokat azonosítva a rendelkezésre álló enrgia táblázatok segítségével megválaszolható a kérdés, hogy az adott elem kimutathatósági határánál nagyobb koncentrációban milyen elemek vannak a felületen.
A SAM berendezésekkel vizsgálható és megjeleníthető egy adott elem felületi eloszlásképe is, ami különösen korróziós, katalitikus vagy szemcsehatár vizsgálatoknál nagy jelentőségű. Egy másik fontos információ egy adott elem mélységi eloszlásgörbéjének a felvétele, amire az AES berendezés porlasztásos üzemmódja ad lehetőséget. Ez főleg többrétegű szerkezeteknél, szegregációs vagy diffúziós vizsgálatoknál fontos.
Kvantitatív analízis
Egy AES mérésből nyerhető közvetlen információ a detektált elemek Auger elektron árama. Az i. elem WXY Auger átmeneténél ez az Auger elektronáram (vagy intenzitás) a következő egyszerűsített alakban adható meg:
![\[I_i(WXZ) {{=}} I_p \cdot P_i(WXY) \cdot \sigma_i(E_p,E_W) \cdot \lambda(E_A) \cdot R(E_p) \cdot T(E_A) \cdot N \cdot X_i\]](/images/math/0/3/a/03a05d587c9466063f6546ffe4225c19.png)
ahol 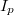 a primer elektronáram erőssége és
a primer elektronáram erőssége és 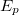 a primer elektronok energiája,
a primer elektronok energiája, 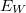 és
és 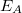 a W atomi nívó ionizációs energiája illetve az Auger energia (az adott Auger átmenetből származó elektronok kinetikus energiája).
a W atomi nívó ionizációs energiája illetve az Auger energia (az adott Auger átmenetből származó elektronok kinetikus energiája). 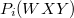 annak a valószínűsége, hogy a gerjesztett atomnak a W-héj ionizációja után WXY típusú Auger-folyamata következik be,
annak a valószínűsége, hogy a gerjesztett atomnak a W-héj ionizációja után WXY típusú Auger-folyamata következik be, 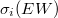 a W-héj ionizációs hatáskeresztmetszete,
a W-héj ionizációs hatáskeresztmetszete, 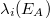 az i. elemből kilépő elektron közepes szabad úthossza,
az i. elemből kilépő elektron közepes szabad úthossza, 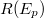 az elektron visszaszórási tényező,
az elektron visszaszórási tényező, 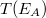 a berendezés transzmissziós együtthatója,
a berendezés transzmissziós együtthatója, 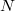 az analizált anyagban lévő atomsűrűség és
az analizált anyagban lévő atomsűrűség és 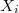 az i. elem koncentrációja (atomtörtben).
Megjegyezzük, hogy a kémiai környezet hatását beleértettük az egyes paraméterek értékeibe. Általában elmondható, hogy a fenti összefüggésben szereplő tényezők a kísérleti körülményeknek és a mátrix-környezetnek bonyolult függvényei, és a mennyiségek utáni zárójelekben csak a legfontosabb paraméterek szerepelnek azok közül, amelyektől ezek a mennyiségek függenek.
az i. elem koncentrációja (atomtörtben).
Megjegyezzük, hogy a kémiai környezet hatását beleértettük az egyes paraméterek értékeibe. Általában elmondható, hogy a fenti összefüggésben szereplő tényezők a kísérleti körülményeknek és a mátrix-környezetnek bonyolult függvényei, és a mennyiségek utáni zárójelekben csak a legfontosabb paraméterek szerepelnek azok közül, amelyektől ezek a mennyiségek függenek.
A mérésekből nyerhető egyik legfontosabb információ az Xi elemkoncentráció. Ennek megállapítására az ad lehetőséget, hogy a (differenciális) energiaspektrum Auger-csúcsainak csúcstól-csúcsig mért nagysága jó közelítéssel arányos az Auger elektronok intenzitásával, ezzel viszont arányos az illető elem koncentrációja. A fenti összefüggésben célszerű bevezetni si érzékenységi faktornak nevezett mennyiséget, ekkor:
![\[I_i(WXY) = I_p \cdot s_i \cdot N \cdot X_i\]](/images/math/6/8/e/68e29c318a21831af705784ea2480dc4.png)
Mint láttuk si nagyon sok, néha csak nagy bizonytalansággal számolható, mennyiségtől függ. Azonban néhány megszorító feltételezés bevezetésével bizonyos mintatípusoknál a mennyiségi meghatározás módja leegyszerűsíthető. Így eltekintünk a felületi érdrsség befolyásoló szerepétől. (Az ebből származó hibát polírozott, vagy sík szemcsehatár felületek előállításával, illetőleg forgatható mintatartó alkalmazásával lehet csökkenteni.) Továbbá figyelembe vesszük, hogy a nem kifejezetten kémiai vegyületeknél a mátrix környezet elsősorban az elemek legkülső elektronhéjainak energiaviszonyait befolyásolja, és a mélynívókra gyakorolt hatása sokkal kisebb jelentőségű. (Így a kiértékelésnél csak azokat az energia csúcsokat használjuk fel, amelyeknek kialakításában a mélynívók vesznek részt.). Az si mátrix függése még tovább csökkenthető, ha relatív érzékenységi tényezőkkel (RSF) dolgozunk, azaz egy önkényesen megválasztott anyag (általában ezüst) érzékenységi faktorához viszonyítjuk (Si=si/sAg). Egy adott berendezés típusra valamennyi tiszta anyag relatív érzékenységi tényezője kimérhető és a fent ismertetett feltételek teljesülése esetén jó közelítéssel használható ismeretlen összetételű anyagokra is. (1) összefüggésből az adott elem koncentrációja (atomtörtben) kifejezve:
![\[X_i {{=}} \frac{I_i(WXY)/s_i}{\sum_{k} I_k(WXY)/s_k}\]](/images/math/8/3/f/83fad0fa413b27d5b5f1406eb5569c15.png)
ahol a k szerinti összegzés a minta összes detektálható elemére terjed ki. Végigosztva a számlálót is és a nevezőt is sAg-vel:
![\[X_i {{=}} \frac{I_i(WXY)/S_i}{\sum_{k} I_k(WXY)/S_k}\]](/images/math/6/4/8/648f6f170607368cc38c8b425727d9da.png)
Ez az összefüggés 10-15% pontossággal alkalmazható nem kémiai vegyületek összetételének meghatározására. A módszer érzékenysége 1-5 at%, ami egyéb kémiai analízissel összehasonlítva igen szerénynek tűnhet. Azonban vegyük figyelembe, hogy az a térfogat amiből az információ származik néhány tized nm vastag réteg, közepesen jól fókuszált primer elektronnyaláb (átmérő: 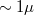 esetében, ~10-8 cm2 területű tartományából. Az atomi sűrűséget 1023 atom/cm3-nek véve 1% érzékenységgel számolva ez néhányszor 104 atom kimutatását jelenti.
esetében, ~10-8 cm2 területű tartományából. Az atomi sűrűséget 1023 atom/cm3-nek véve 1% érzékenységgel számolva ez néhányszor 104 atom kimutatását jelenti.
Kötésállapot kimutatása
Az Auger mérésekből nyerhető másik fontos információ a kémiai kötésviszonyokra levonható következtetés. Erre az ad lehetőséget, hogy a különböző kémiai környezet
Mérési feladatok
Ötvözet-minta összetételének meghatárzása
A méréshez szükséges eszközök:
- acél-etalon minta (a mérés kezdetén már a vákuumkamra mintatartójára helyezve),
- SAM berendezés,
- Vákuummérő,
- Dewar-palack cseppfolyós nitrogénnel töltve.
Az előzőekben már láttuk, hogy az Auger-elektronok intenzitását számos mátrixfüggő és berendezésfüggő paraméter befolyásolhatja. Ezek közül sok paraméternek a hatásától eltekinthetünk, ha néhány egyszerűsítő körülménnyel és feltételezéssel élünk. Az első az, hogy az információs mélységen belül eltekintünk ugyanazon elem koncentrációbeli inhomogenitásaitól, tehát megelégszünk azzal, hogy a legfelső atomrétegek átlagos összetételét határozzuk meg. Ugyanakkor laterális vonatkozásban, a primer gerjesztő nyaláb keresztmetszetének megfelelő területen belül szintén eltekintünk a minta alkotóinak koncentrációbeli inhomogenitásaitól, tehát e tekintetben is csak átlagértéket határozunk meg. A következő egyszerűsítés az, hogy eltekintünk a felületi érdességi faktor szerepétől. Ezt azért vagyunk kénytelenek megtenni, mert a mérés (és a porlasztás) alatt egyszerűen nincs idő a minta egyedi felületi geometriai sajátságainak pontos feltérképezésére. Az ebből származó hibát úgy csökkentjük, hogy simára polírozott felületeket, vagy a gerjesztő elektronnyaláb alatt síknak tekinthető szemcsehatárokat vizsgálunk.
További egyszerűsítésre nyílik lehetőség, ha meggondoljuk, hogy a mátrixkörnyezet elsősorban az elemek legkülső elektronhéjainak energiaviszonyait befolyásolja, és a mélynívókra gyakorolt hatása sokkal kisebb jelentőségű, esetenként elhanyagolható. Így, ha a mennyiségi kiértékelésnél azokat az Auger elektronenergia csúcsokat használjuk fel, amelyeknek a kialakításában a mélynívók vesznek részi, akkor (hacsak nem kémiai vegyületet vizsgálunk) egy hasonló összetételű etalonmintával, vagy csak a vizsgált elemeket tartalmazó tiszta anyaggal történő összehasonlításnál az Auger-folyamat-keltés valószínűségében és az ionizációs hatáskeresztmetszetben fellépő változások elhanyagolhatóak. Továbbá, ha a vizsgált minta-ötvözet a periódusos rendszerben közeli rendszámú fémes elemekből áll, amelyeknél az elektron közepes szabad úthossz és a visszaszórási tényező értékek közel azonosak, akkor a mennyiségi kiértékelésre néhány százalékos relatív hibával alkalmas az un. relatív érzékenységi tényezők módszere. Ennél a módszernél a minták összetételének meghatározására felhasználjuk az ugyanazon típusú berendezésen egy ezüst-etalon mintára normált érzékenységi faktorokat a különböző elemek esetén (SX). Ezek az értékek : 3 keV, 5 keV és 10 keV primer elektronenergiákra a 4., 5. és 6, ábrákról olvashatók le.
Ezután a vizsgált minta x. komponensének koncentrációját a következő összefüggéssel számolhatjuk atomszázalékban:
![\[{{C_x = \frac{I_x}{L_x \cdot E_{mx} \cdot I_{px} \cdot S_x} \bigg / \sum_{i=1}^n \frac {I_x}{L_x \cdot E_{mx} \cdot I_{px} \cdot S_x}} \cdot 100 }\]](/images/math/4/0/0/400829422228e17398dd0ad675bf5c30.png)
ahol
- Ii az i. komponens csúcstól-csúcsig mért Auger-elektron intenzitása
- Si az i. elem relatív érzékenységi tényezője
- Emi az i. elemnél alkalmazott modulációs energia
- Li az i. elemnél alkalmazott erősítési tényező
- Ipi az i. elemnél alkalmazott primer elektron áram
n a minta komponenseinek száma
Ha a mérések folyamán a SAM berendezés erősítési tényezőjét, modulációs energiáját és a primer elektron áramot nem változtatjuk (és a jelen esetben így fogunk eljárni), akkor (5) a következő egyszerűbb alakot ölti:
![\[{{C_x = \frac{I_x}{S_x} \Big / \sum_{I=1}^n\frac{I_x}{S_x} \cdot 100}}\]](/images/math/2/6/1/2613a484b6dfca5bd49cd8bdc126efe8.png)
Ezek után az elvégzendő feladat egy ismeretlen összetételű acélminta komponenseinek kvalitatív, majd mennyiségi meghatározása. A mintának a mintatartóba történő behelyezése után, és a vákuumszivattyúk elindítását követően legalább egy nappal később kerüljön sor a mérésre a megfelelő nagyvákuum biztosítása céljából. A mintabetétel és a vákuumszivattyúk bekapcsolása a mérés kezdetére megtörtént. Közvetlenül a mérések megkezdése előtt a vákuumrendszer falába történő cseppfolyós levegő beengedésével javítsuk tovább a nagyvákuum értéket 10-7-10-8 Pa nyomás eléréséig. A vákuumszivattyúkat szabályozó egység a 7. ábrán látható 12. fiókban nyert elhelyezést.
Magát a mérést azzal kezdjük, hogy a mintát geometriailag optimális pozícióba hozzuk a 2000 eV-os rugalmasan szórt energiacsúcs mérésével. Ezt a mérés kezdetén a felügyelő tanár segítségével végezzük el. Ezután a 7. ábrán látható SAM berendezés 8., 10. és 11. fiókjának és a készülék TV¬monitorának képernyőjén az abszorbeált áramképek segítségével kiválasztjuk a vizsgálatokra legalkalmasabb homogén felületrészt, ami kiválásokat, inhomogenitásokat nem tartalmaz. A mérési hely pontos beállítását a SAM berendezésen a 8. fiókon a kép-pozícionáló egység X-Y jelzésű potenciométereivel végezhetjük.
Ezután a 7. jelű ionágyú-szabályozó egység alkalmazásával ionporlasztással megtisztítjuk a mintafelületet. Az ionporlasztást addig alkalmazzuk, amíg a mintafelületről származó szén- és oxigén-csúcsok eltűnnek, vagy minimális értéken stabilizálódnak. Az ion- és elektronáramot a 9. fiók digitális kijelzőjén olvashatjuk le.
Ezt követően kerül sor a minta Auger-elektronenergia spektrumának felvételére, célszerűen 5 keV-es primer elektronnyaláb alkalmazásával. A primer energiát a 11. fiók BEAM VOLTAGE feliratú potenciométerével állítjuk be. Az erősítési tényező és elektronsokszorozó nagyfeszültség értékeket úgy állítjuk be, hogy az Auger-energiacsúcsok legnagyobbika függőleges irányban közel kitöltse az írószerkezetre helyezett spektrumpapírt.
A primer elektronáram értékét 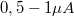 között állítsuk be. Az erősítési tényezőt az 5. fiókon, az elektronsokszorozó nagyfeszültséget a 6. fiókon állítjuk be. A primer elektronáramot a 10. fiók EMISSION és CONDENSER LENS feliratú potenciométereivel állítjuk be, és a 9. fiók digitális kijelzőjén olvassuk le.
között állítsuk be. Az erősítési tényezőt az 5. fiókon, az elektronsokszorozó nagyfeszültséget a 6. fiókon állítjuk be. A primer elektronáramot a 10. fiók EMISSION és CONDENSER LENS feliratú potenciométereivel állítjuk be, és a 9. fiók digitális kijelzőjén olvassuk le.
A spektrumfelvételt a 4. fiók START feliratú gombjának benyomásával indítjuk, mellyel egyidőben a 2. fiók írószerkezetén a tollat ráhelyezzük a spektrumpapírra. A folyamatosan, kb. 5 perc alatt kiíródó spektrum megjelenik az 1. fiók oszcilloszkópján is. A 3. fiókot, ami mélységi profilgörbe felvételét teszi lehetővé, ennél a mérésnél nem használjuk. Az aktuális Auger-elektronenergia értékeket a 4. fiók digitális kijelzőjén olvashatjuk le. A spektrumot elég a 0-1000 eV-os energiaintervallumban felvenni.
A spektrum felvétele után először a 2. ábrán látható Auger-elektronenergia táblázat felhasználásával azonosítjuk a mintát alkotó elemeket. Az azonosításnál célszerű annak figyelembevétele, hagy a fémek többségének több, karakterisztikus energiájú Auger-energiacsúcsa is van. A beazonosított elemek vegyjelét ceruzával írjuk rá a spektrumon lévő és az adott elemet reprezentáló csúcsra.
A mérési feladat második részében először olvassuk le mm-ben a beazonosított fém-komponensek fő Auger-csúcsainak csúcstól-csúcsig mért intenzitását. Ennek ismeretében, továbbá az 5. ábra és a (6) összefüggés segítségével határozzuk meg mennyiségileg is a minta összetételét. A mennyiségi meghatározás eredményét táblázatosan foglaljuk össze.
Kémiai eltolódás (chemical shift) vizsgálata
A méréshez szükséges eszközök:
- 1 db szilícium-szilíciumdioxid minta ,
- AES berendezés,
- Vákuummérő,
- Dewar-palack cseppfolyós nitrogénnel töltve.
Az előzőekben már volt arról szó, hogy a különböző kémiai környezet különbező mértékű eltolódásokat hozhat létre az Auger-elektronenergia csúcsok helyében. Ez az eltolódás különösen szignifikáns a kis energiájú Auger-elektronenergia csúcsoknál, amelyekhez tartozó átmenetekben a vegyértéksáv is részt vesz.
A kémiai eltolódás jól tanulmányozható a tiszta Si, illetve a SiO2 minták Auger - elektronenergia spektrumainak összehasonlításával.
A mérés kivitelezéséhez egy Si-hordozóra növesztett SiO2 réteget tartalmazó mintát helyezünk a SAM berendezés mintatartójába. Ez a mérés kezdetére már megtörtént. A mérések megkezdése előtt a megfelelő nagyvákuum biztosítása céljából az ezzel kapcsolatban a 4.1. pontban leírt műveleteket végezzük el. A mintát a mérés előtt geometriailag optimális pozícióba állítjuk a 2000eV-os rugalmasan szórt energiacsúcs mérésével.
Ezt követően a mintán az abszorbeált áram-kép megjelenítésével kiválasztjuk az optimális mérési helyet, a 4.1. pontban leírt módon. Ezután a 4.1. pontban leírt módon itt is alkalmazzunk egy rövid idejű ionporlasztást a felület tisztítása céljából.
Az ionporlasztással letisztított SiO2 réteg felületéről vegyünk fel egy Auger- elektronenergia spektrumot 5 keV primer elektronenergia alkalmazásával, melyet a 11. fiókon állítunk be. A primer elektronáram néhány tized mikroamper legyen. A spektrumfelvétel folyamata a 4.1. pontban leírtak szerint történjen, azzal a különbséggel, hogy ennél a mérésnél célszerű az energiaspektrumot a 0-100 eV-os intervallumban felvenni, amelyet a 4. fiók SCALE DIVISION potenciométerének 10-es állásba tevésével érhetünk el.
Ezután a mintát addig porlasztjuk az Ar-ionokkal, amíg teljesen leporlódik a SiO2 réteg, és elérjük a tiszta Si-hordozót. Ennek a folyamatnak a nyomon követésére a rétegporlasztás elején állítsuk be az oxigén Auger elektron-csúcsát az 1. fiók oszcilloszkópján és a porlasztást addig folytassuk, amíg az oxigén csúcsa eltűnik. Ezután a tiszta Si-rétegben ismét vegyünk fel a 0-100 eV-os intervallumban egy Auger-elektronenergia spektrumot. Ha az itt leírt mérésekhez változtatni kell az erősítési tényező, illetve az elektronsokszorozó nagyfeszültség-értékeket, akkor ezt az 5. fiók SENSITIVITY feliratú gombjának, illetve a 6. fiók potenciométereinek változtatásával érhetjük el.
A SiO2, illetve a Si rétegeken az előbbi módon felvett Auger-elektronenergia spektrumokon a vízszintes energia-tengelyen leolvassuk a szilícium kisenergiás, LMM átmenethez tartozó Auger-csúcsának a helyét. A két mintánál így megállapított energiaértékek különbségéből meghatározzuk a kémiai eltolódás mértékét.
Javasolt irodalom
- Gergely Gy: Szekunder emissziós spektrometria, AES, SEES, ELS. A szilárdtestkutatás újabb eredményei, Akadémia Kiadó, Budapest, 1979.
- Szilárd testek vizsgálata elektronokkal, ionokkal és röntgensugárzással. Szerkesztette: 4. Brümmer, J. Heydenreich, K.N. Krebs, N.G. Schneider. Műszaki Könyvkiadó, Budapest, 1984.
- Thompson, M.D. Baker, A. Christie, J.F. Tyson: Auger Electron Spectroscopy. John Wiley and Sons. N.Y.1985-86.
PDF formátum